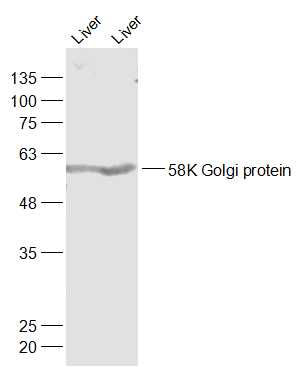
产品货号 : mlR23473
英文名称 : 58K Golgi protein
中文名称 : 58K高尔基蛋白抗体
别 名 : Formimidoyltetrahydrofolate cyclodeaminase; Formimidoyltransferase cyclodeaminase; Formiminotetrahydrofolate cyclodeaminase; Formiminotransferase cyclodeaminase; Formiminotransferase-cyclodeaminase; FTCD; FTCD_HUMAN; Glutamate formiminotransferase; Glutamate formyltransferase; LCHC 1; LCHC1.
研究领域 : 肿瘤 信号转导
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Pig, Cow, Sheep,
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 59kDa
细胞定位 : 细胞浆
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human 58K Golgi protein :151-250/541
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : The protein encoded by this gene is a bifunctional enzyme that channels 1-carbon units from formiminoglutamate, a metabolite of the histidine degradation pathway, to the folate pool. Mutations in this gene are associated with glutamate formiminotransferase deficiency. Alternatively spliced transcript variants have been found for this gene.[provided by RefSeq, Dec 2009]
Function:
Folate-dependent enzyme, that displays both transferase and deaminase activity. Serves to channel one-carbon units from formiminoglutamate to the folate pool. Binds and promotes bundling of vimentin filaments originating from the Golgi.
Subcellular Location:
Cytoplasm, cytoskeleton, centrosome, centriole. Golgi apparatus. More abundantly located around the mother centriole.
DISEASE:
Defects in FTCD are the cause of glutamate formiminotransferase deficiency (FIGLU-URIA) [MIM:229100]; also known as formiminoglutamicaciduria (FIGLU-uria). It is an autosomal recessive disorder. Features of a severe phenotype, include elevated levels of formiminoglutamate (FIGLU) in the urine in response to histidine administration, megaloblastic anemia, and mental retardation. Features of a mild phenotype include high urinary excretion of FIGLU in the absence of histidine administration, mild developmental delay, and no hematological abnormalities.
Similarity:
In the C-terminal section; belongs to the cyclodeaminase/cyclohydrolase family.In the C-terminal section; belongs to the cyclodeaminase/cyclohydrolase family. In the N-terminal section; belongs to the formiminotransferase family. In the N-terminal section; belongs to the formiminotransferase family.
SWISS:
O95954
Gene ID:
10841
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
产品图片