产品货号 : mlR6311
英文名称 : BSCL2
中文名称 : 先天性脂肪代谢障碍蛋白2抗体(常染色体显性遗传痉挛性截瘫17)
别 名 : Bernardinelli Seip congenital lipodystrophy 2; Bernardinelli Seip congenital lipodystrophy type 2 protein; Bernardinelli-Seip congenital lipodystrophy type 2 protein; BSCL 2; BSCL2; BSCL2_HUMAN; GNG3LG; HMN 5; HMN5; MGC4694; Seipin; Spastic paraplegia 17 (autosomal dominant); Spastic paraplegia 17 (Silver syndrome); Spastic paraplegia 17; Spastic paraplegia with amyotrophy of hands and feet (Silver syndrome); Spastic paraplegia with amyotrophy of hands and feet; SPG 17; SPG17.
研究领域 : 心血管 细胞生物 免疫学 神经生物学 细胞类型标志物
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Dog, Cow, Horse, Rabbit,
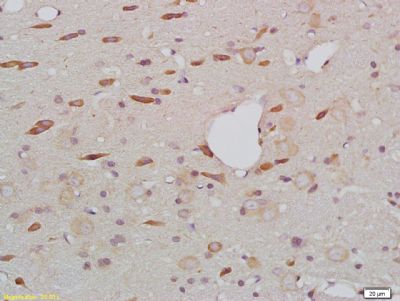
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 44kDa
细胞定位 : 细胞浆
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human BSCL2/SPG17:151-250/398
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : Defects in BSCL2 are the cause of congenital generalized lipodystrophy type 2 (CGL2) . Congenital generalized lipodystrophy is an autosomal recessive disorder characterized by a near absence of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes.
Defects in BSCL2 are the cause of spastic paraplegia type 17 (SPG17) ; also known as Silver spastic paraplegia syndrome. Spastic paraplegia is a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. SPG17 is characterized by prominent amyotrophy of the hand muscles, the presence of mild to severe pyramidal tract signs, and spastic paraplegia.
SPG17 is a motor neuron disease overlapping with distal spinal muscular atrophy type 5. Defects in BSCL2 are a cause of distal hereditary motor neuropathy type 5 (HMN5); also known aS distal hereditary motor neuropathy type V (DSMAV). HMN5 is an autosomal dominant disorder characterized by degeneration of motor nerve fibers, predominantly in limb distal regions.
Function:
Endoplasmic reticulum membrane; Multi-pass membrane protein.
Tissue Specificity:
Highest expression in brain and testis.
DISEASE:
Defects in BSCL2 are the cause of congenital generalized lipodystrophy type 2 (CGL2) [MIM:269700]. Congenital generalized lipodystrophy is an autosomal recessive disorder characterized by a near absence of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes.
Defects in BSCL2 are the cause of spastic paraplegia type 17 (SPG17) [MIM:270685]; also known as Silver spastic paraplegia syndrome. Spastic paraplegia is a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. SPG17 is characterized by prominent amyotrophy of the hand muscles, the presence of mild to severe pyramidal tract signs, and spastic paraplegia. SPG17 is a motor neuron disease overlapping with distal spinal muscular atrophy type 5.
Defects in BSCL2 are a cause of distal hereditary motor neuropathy type 5 (HMN5) [MIM:600794]; also known aS distal hereditary motor neuropathy type V (DSMAV). HMN5 is an autosomal dominant disorder characterized by degeneration of motor nerve fibers, predominantly in limb distal regions.
Similarity:
Belongs to the seipin family.
SWISS:
Q96G97
Gene ID:
26580
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
产品图片