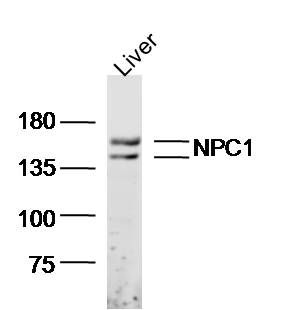
产品货号 : mlR6764
英文名称 : NPC1
中文名称 : 尼曼匹克C1前体蛋白抗体
别 名 : Niemann Pick C1; Niemann Pick C1 protein precursor; Niemann Pick disease, type C1; Niemann-Pick C1 protein; NPC; NPC1; NPC1_HUMAN.
研究领域 : 心血管 细胞生物 神经生物学
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Chicken, Pig, Guinea Pig,
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 138kDa
细胞定位 : 细胞浆 细胞膜
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from mo NPC1/Niemann Pick C1:1181-1278/1287
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : This gene encodes a large protein that resides in the limiting membrane of endosomes and lysosomes and mediates intracellular cholesterol trafficking via binding of cholesterol to its N-terminal domain. It is predicted to have a cytoplasmic C-terminus, 13 transmembrane domains, and 3 large loops in the lumen of the endosome - the last loop being at the N-terminus. This protein transports low-density lipoproteins to late endosomal/lysosomal compartments where they are hydrolized and released as free cholesterol. Defects in this gene cause Niemann-Pick type C disease, a rare autosomal recessive neurodegenerative disorder characterized by over accumulation of cholesterol and glycosphingolipids in late endosomal/lysosomal compartments.[provided by RefSeq, Aug 2009].
Function:
Involved in the intracellular trafficking of cholesterol. May play a role in vesicular trafficking in glia, a process that may be crucial for maintaining the structural and functional integrity of nerve terminals.
Subunit:
Interacts with TMEM97.
Subcellular Location:
Late endosome membrane; Multi-pass membrane protein. Lysosome membrane; Multi-pass membrane protein.
Post-translational modifications:
Glycosylated.
DISEASE:
Defects in NPC1 are the cause of Niemann-Pick disease type C1 (NPC1) [MIM:257220]. A lysosomal storage disorder that affects the viscera and the central nervous system. It is due to defective intracellular processing and transport of low-density lipoprotein derived cholesterol. It causes accumulation of cholesterol in lysosomes, with delayed induction of cholesterol homeostatic reactions. Niemann-Pick disease type C1 has a highly variable clinical phenotype. Clinical features include variable hepatosplenomegaly and severe progressive neurological dysfunction such as ataxia, dystonia and dementia. The age of onset can vary from infancy to late adulthood. An allelic variant of Niemann-Pick disease type C1 is found in people with Nova Scotia ancestry. Patients with the Nova Scotian clinical variant are less severely affected.
Similarity:
Belongs to the patched family.
Contains 1 SSD (sterol-sensing) domain.
SWISS:
O15118
Gene ID:
4864
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
尼曼匹克病-神经磷沉积性疾病,主要是由于神经磷脂酶(sphingomyelinase)缺乏所致,神经鞘磷脂酶(sphingomyelinase)缺乏致神经鞘磷脂代谢障碍。导致后者蓄积在单核巨噬细胞系统内,出现肝、脾肿大,中枢神经系统退行性变。神经鞘磷脂是由N-酰鞘氨醇与一个分子的磷酸胆硷(phosphocholine)在C1、部位连接而成,神经鞘磷脂来源于各种细胞膜和红细胞基质等。在细胞代谢衰老过程中被巨噬细胞吞噬,神经磷脂酶缺少后,全身神经鞘磷脂代谢紊乱,神经磷脂沉积在单核-巨噬细胞系统和神经组织细胞中。
产品图片