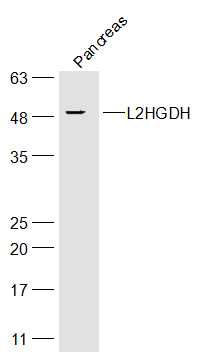
产品货号 : mlR16876
英文名称 : L2HGDH
中文名称 : L2HGDH蛋白抗体
别 名 : 2 hydroxyglutarate dehydrogenase; Alpha hydroxyglutarate oxidoreductase; Alpha ketoglutarate reductase; C14orf160; Duranin; FLJ12618; L alpha hydroxyglutarate dehydrogenase; L-2-hydroxyglutarate dehydrogenase; L-2-hydroxyglutarate dehydrogenase, mitochondrial; L2HDH_HUMAN; l2hgdh; mitochondrial.
研究领域 : 细胞生物 神经生物学 信号转导 新陈代谢
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Pig, Cow, Horse, Rabbit, Sheep, Orangutan
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 45kDa
细胞定位 : 细胞浆
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human L2HGDH:201-300/463
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : This gene encodes L-2-hydroxyglutarate dehydrogenase, a FAD-dependent enzyme that oxidizes L-2-hydroxyglutarate to alpha-ketoglutarate in a variety of mammalian tissues. Mutations in this gene cause L-2-hydroxyglutaric aciduria, a rare autosomal recessive neurometabolic disorder resulting in moderate to severe mental retardation. [provided by RefSeq, Jul 2008]
Subcellular Location:
Mitochondrion.
Tissue Specificity:
Widely expressed. Highly expressed in brain, testis and muscle. Expressed to a lower extent in lymphocytes, fibroblasts, keratinocytes, placenta, bladder, small intestine, liver and bone marrow.
DISEASE:
Defects in L2HGDH are the cause of L-2-hydroxyglutaric aciduria (L2HGA) [MIM:236792]. L2HGA is a rare autosomal recessive disorder clinically characterized by mild psychomotor delay in the first years of life, followed by progressive cerebellar ataxia, dysarthria and moderate to severe mental retardation. Diagnosis is based on the presence of an excess of L-2-hydroxyglutaric acid in urine, blood and cerebrospinal fluid.
Similarity:
Belongs to the L2HGDH family.
SWISS:
Q9H9P8
Gene ID:
79944
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
产品图片