产品货号 : mlR33203
英文名称 : HSP27
中文名称 : 热休克蛋白27单克隆抗体
别 名 : Heat shock 27kDa protein; 28 kDa heat shock protein; CMT2F; DKFZp586P1322; Estrogen regulated 24 kDa protein; Estrogen-regulated 24 kDa protein; Heat shock 25kDa protein 1; Heat shock 25kDa protein 1; Heat shock 27 kDa protein; Heat shock 27kD protein 1; Heat shock 27kDa protein 1; Heat shock 27kDa protein 1; Heat shock 28kDa protein 1; Heat shock 28kDa protein 1; Heat Shock Protein 27; Heat Shock Protein 27; Heat shock protein beta 1; Heat shock protein beta-1; Heat Shock Protein27; Heat Shock Protein27; HMN2B; HS.76067; Hsp 25; Hsp 25; Hsp 27; Hsp 27; Hsp 28; Hsp 28; Hsp B1; Hsp B1; Hsp25; Hsp25; HSP27; Hsp28; Hsp28; HspB1; HspB1; HSPB1_HUMAN; SRP27; Stress responsive protein 27; Stress-responsive protein 27.
研究领域 : 肿瘤 免疫学 信号转导
抗体来源 : Mouse
克隆类型 : Monoclonal
克 隆 号 : 1G12
交叉反应 : Human,
产品应用 : WB=1:1000-2000
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 27kDa
细胞定位 : 细胞核 细胞浆
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : Recombinant human HSP27 Protein:
亚 型 : IgG
纯化方法 : affinity purified by Protein G
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : The protein encoded by this gene is induced by environmental stress and developmental changes. The encoded protein is involved in stress resistance and actin organization and translocates from the cytoplasm to the nucleus upon stress induction. Defects in this gene are a cause of Charcot-Marie-Tooth disease type 2F (CMT2F) and distal hereditary motor neuropathy (dHMN). [provided by RefSeq, Oct 2008]
Function:
Involved in stress resistance and actin organization.
Subunit:
Interacts with TGFB1I1. Associates with alpha- and beta-tubulin, microtubules and CRYAB. Interacts with HSPB8 and HSPBAP1.
Subcellular Location:
Cytoplasm. Nucleus. Cytoplasm, cytoskeleton, spindle. Note=Cytoplasmic in interphase cells. Colocalizes with mitotic spindles in mitotic cells. Translocates to the nucleus during heat shock and resides in sub-nuclear structures known as SC35 speckles or nuclear splicing speckles.
Tissue Specificity:
Detected in all tissues tested: skeletal muscle, heart, aorta, large intestine, small intestine, stomach, esophagus, bladder, adrenal gland, thyroid, pancreas, testis, adipose tissue, kidney, liver, spleen, cerebral cortex, blood serum and cerebrospinal fluid. Highest levels are found in the heart and in tissues composed of striated and smooth muscle.
Post-translational modifications:
Phosphorylated in MCF-7 cells on exposure to protein kinase C activators and heat shock.
Phosphorylation by MAPKAPK2 and MAPKAPK3 in response to stress leads to dissociate HSP27/HSPB1 from large small heat-shock protein (sHsps) oligomers and impair its chaperone activity and ability to protect against oxidative stress effectively. Phosphorylation by MAPKAPK5 in response to PKA stimulation induces F-actin rearrangement.
DISEASE:
Defects in HSPB1 are the cause of Charcot-Marie-Tooth disease type 2F (CMT2F) [MIM:606595]. CMT2F is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Neuropathies of the CMT2 group are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. Nerve conduction velocities are normal or slightly reduced. CMT2F onset is between 15 and 25 years with muscle weakness and atrophy usually beginning in feet and legs (peroneal distribution). Upper limb involvement occurs later. CMT2F inheritance is autosomal dominant.
Defects in HSPB1 are a cause of distal hereditary motor neuronopathy type 2B (HMN2B) [MIM:608634]. Distal hereditary motor neuronopathies constitute a heterogeneous group of neuromuscular disorders caused by selective impairment of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.
Similarity:
Belongs to the small heat shock protein (HSP20) family.
SWISS:
P04792
Gene ID:
3315
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
信号传导(Signaling Intermediates)
适用组织:石蜡切片 细胞定位:胞浆和部分胞核
HSPs是细胞受应激原刺激后诱导产生的一组应激蛋白,与肿瘤发生、 增殖及分化有关。按其分子量不同可分为3种类型,每组的HSPs的分布及功能有所不同。 热休克蛋白27是人体中最常见而又最小的热休克蛋白。HSP27和其它HSPs可能与肿瘤耐药和肿瘤的分化程度以及病人的预后有关。
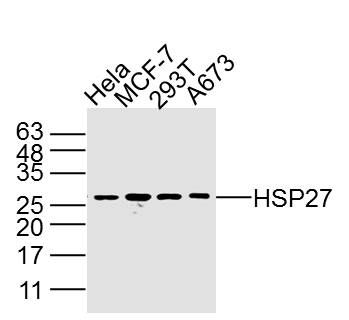
产品图片