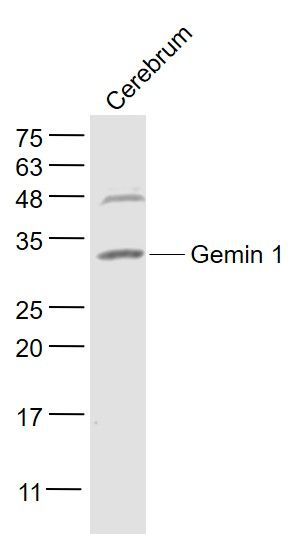
产品货号 : mlR11561
英文名称 : Gemin 1
中文名称 : 脊髓性肌萎缩症蛋白SMA/抗体
别 名 : Component of gems 1; Component of gems 2; Gemin 1; Gemin-1; SMA; SMA1; SMA3; SMN; SMN_HUMAN; SMN1; SMN2; SMNC; SMNT; Survival motor neuron protein; survival of motor neuron 1, telomeric; survival of motor neuron 2, centromeric;Component of gems 1; Gemin 1; Gemin-1; Gemin1; SMA 1; SMA 2; SMA 3; SMA 4; SMA; SMA1; SMA2; SMA3; SMA4; SMN 1; SMN; SMN-1; SMN_HUMAN; SMN1; SMN2; SMNT; Survival motor neuron protein; Survival of motor neuron 1 (telomeric); survival of motor neuron 1; Survival of motor neuron 1, telomeric; T-BCD541; BCD541; SMN_HUMAN.
研究领域 : 细胞生物 神经生物学 表观遗传学
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Dog, Pig, Cow, Rabbit,
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 31kDa
细胞定位 : 细胞核 细胞浆
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human Gemin 1:31-100/294
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : This gene is part of a 500 kb inverted duplication on chromosome 5q13. This duplicated region contains at least four genes and repetitive elements which make it prone to rearrangements and deletions. The repetitiveness and complexity of the sequence have also caused difficulty in determining the organization of this genomic region. The telomeric and centromeric copies of this gene are nearly identical and encode the same protein. However, mutations in this gene, the telomeric copy, are associated with spinal muscular atrophy; mutations in the centromeric copy do not lead to disease. The centromeric copy may be a modifier of disease caused by mutation in the telomeric copy. The critical sequence difference between the two genes is a single nucleotide in exon 7, which is thought to be an exon splice enhancer. Note that the nine exons of both the telomeric and centromeric copies are designated historically as exon 1, 2a, 2b, and 3-8. It is thought that gene conversion events may involve the two genes, leading to varying copy numbers of each gene. The protein encoded by this gene localizes to both the cytoplasm and the nucleus. Within the nucleus, the protein localizes to subnuclear bodies called gems which are found near coiled bodies containing high concentrations of small ribonucleoproteins (snRNPs). This protein forms heteromeric complexes with proteins such as SIP1 and GEMIN4, and also interacts with several proteins known to be involved in the biogenesis of snRNPs, such as hnRNP U protein and the small nucleolar RNA binding protein. Two transcript variants encoding distinct isoforms have been described. [provided by RefSeq, Sep 2008]
Function:
The SMN complex plays an essential role in spliceosomal snRNP assembly in the cytoplasm and is required for pre-mRNA splicing in the nucleus. It may also play a role in the metabolism of snoRNPs.
Subunit:
Component of an import snRNP complex composed of KPNB1, RNUT1, SMN1 and ZNF259. Part of the core SMN complex that contains SMN1, GEMIN2/SIP1, DDX20/GEMIN3, GEMIN4, GEMIN5, GEMIN6, GEMIN7, GEMIN8 and STRAP/UNRIP. Interacts with DDX20, FBL, NOLA1, RNUT1, SYNCRIP and with several spliceosomal snRNP core Sm proteins, including SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE and ILF3. Interacts with OSTF1, LSM10 and LSM11.
Subcellular Location:
Cytoplasm. Nucleus, gem. Note=Localized in subnuclear structures next to coiled bodies, called Gemini of Cajal bodies (Gems).
Tissue Specificity:
Expressed in a wide variety of tissues. Expressed at high levels in brain, kidney and liver, moderate levels in skeletal and cardiac muscle, and low levels in fibroblasts and lymphocytes. Also seen at high levels in spinal cord. Present in osteoclasts and mononuclear cells (at protein level).
DISEASE:
Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 1 (SMA1) [MIM:253300]. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Autosomal recessive forms are classified according to the age of onset, the maximum muscular activity achieved, and survivorship. The severity of the disease is mainly determined by the copy number of SMN2, a copy gene which predominantly produces exon 7-skipped transcripts and only low amount of full-length transcripts that encode for a protein identical to SMN1. Only about 4% of SMA patients bear one SMN1 copy with an intragenic mutation. SMA1 is a severe form, with onset before 6 months of age. SMA1 patients never achieve the ability to sit.
Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 2 (SMA2) [MIM:253550]. SMA2 is an autosomal recessive spinal muscular atrophy of intermediate severity, with onset between 6 and 18 months. Patients do not reach the motor milestone of standing, and survive into adulthood. Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 3 (SMA3) [MIM:253400]. SMA3 is an autosomal recessive spinal muscular atrophy with onset after 18 months. SMA3 patients develop ability to stand and walk and survive into adulthood. Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 4 (SMA4) [MIM:271150]. SMA4 is an autosomal recessive spinal muscular atrophy characterized by symmetric proximal muscle weakness with onset in adulthood and slow disease progression. SMA4 patients can stand and walk.
Similarity:
Belongs to the SMN family.
Contains 1 Tudor domain.
SWISS:
Q16637
Gene ID:
6606
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
产品图片