产品货号 : mlR6292
英文名称 : Bone Alkaline Phosphatase
中文名称 : 骨碱性磷酸酶抗体
别 名 : AKP2; Alkaline phosphatase liver/bone/kidney; Alkaline phosphatase liver/bone/kidney isozyme; Alkaline phosphatase tissue nonspecific isozyme; Alkaline phosphatase, tissue-nonspecific isozyme; ALPL; AP TNAP; AP-TNAP; APTNAP; BALP; BAP; FLJ40094; FLJ93059; Glycerophosphatase; HOPS; Liver/bone/kidney isozyme; Liver/bone/kidney type alkaline phosphatase; MGC161443; MGC167935; PHOA; PPBT_HUMAN; Tissue non specific alkaline phosphatase; Tissue nonspecific ALP; TNAP; TNSALP.
研究领域 : 肿瘤 细胞生物 免疫学 信号转导 干细胞 激酶和磷酸酶 细胞骨架 细胞外基质
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Cow, Rabbit,
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 Flow-Cyt=1ug/Test IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 55kDa
细胞定位 : 细胞膜
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human Bone Alkaline Phosphatase:56-150/524
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : Defects in ALPL are a cause of hypophosphatasia (HOPS) . HOPS is an inherited metabolic bone disease characterized by defective skeletal mineralization. Four hypophosphatasia forms are distinguished, depending on the age of onset: perinatal, infantile, childhood and adult type. The perinatal form is the most severe and is almost always fatal. Patients with only premature loss of deciduous teeth, but with no bone disease are regarded as having odontohypophosphatasia.
Function:
This isozyme may play a role in skeletal mineralization.
Subunit:
Homodimer.
Subcellular Location:
Cell membrane; Lipid-anchor, GPI-anchor.
Post-translational modifications:
Glycosylated.
DISEASE:
Defects in ALPL are a cause of hypophosphatasia (HOPS) [MIM:146300]. HOPS is an inherited metabolic bone disease characterized by defective skeletal mineralization. Four hypophosphatasia forms are distinguished, depending on the age of onset: perinatal, infantile, childhood and adult type. The perinatal form is the most severe and is almost always fatal. Patients with only premature loss of deciduous teeth, but with no bone disease are regarded as having odontohypophosphatasia (odonto).
Defects in ALPL are a cause of hypophosphatasia childhood type (HOPSC) [MIM:241510].
Defects in ALPL are a cause of hypophosphatasia infantile type (HOPSI) [MIM:241500].
Similarity:
Belongs to the alkaline phosphatase family.
SWISS:
P05186
Gene ID:
249
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
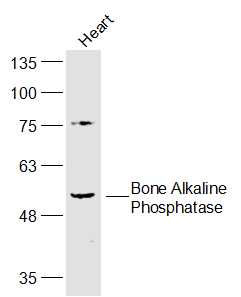
产品图片