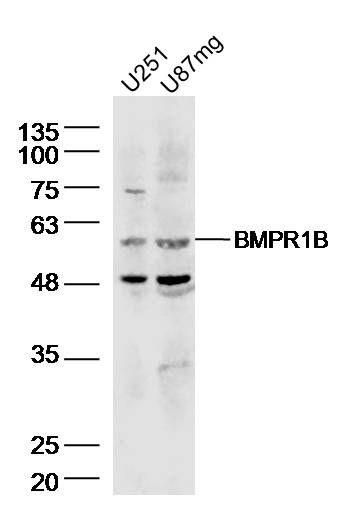
产品货号 : mlR6639
英文名称 : BMPR1B
中文名称 : 骨形态发生蛋白受体1B抗体
别 名 : BMPR-IB; Activin receptor like kinase 6; Acvrlk6; ALK 6; ALK6; alk6tr; BMP type-1B receptor; BMPR IB; BMPR-1B; Bmpr1b; BMPRIB; BMR1B_HUMAN; Bone morphogenetic protein receptor type 1B; Bone morphogenetic protein receptor type IB; Bone morphogenetic protein receptor type-1B; BR 1b; BR1b; CDw 293; CDw293; CDw293 antigen; CFK 43a; CFK43a; Serine/threonine receptor kinase; zALK 6; zALK6.
研究领域 : 细胞生物 信号转导 干细胞 转录调节因子 激酶和磷酸酶 细胞表面分子 细胞外基质 表观遗传学
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Dog, Cow, Rabbit, Sheep, .
产品应用 : WB=1:500-2000 ELISA=1:500-1000
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
分 子 量 : 56kDa
细胞定位 : 细胞膜
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human BMPR1B:61-160/502 <Extracellular>
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍background:
On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. Type II receptors phosphorylate and activate type I receptors which autophosphorylate, then bind and activate SMAD transcriptional regulators. Receptor for BMP7/OP-1 and GDF5.
Involvement in disease; Defects in BMPR1B are the cause of acromesomelic chondrodysplasia with genital anomalies (AMDGA). Acromesomelic chondrodysplasias are rare hereditary skeletal disorders characterized by short stature, very short limbs, and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers).
Defects in BMPR1B are a cause of brachydactyly type A2 (BDA2) [MIM:112600]. Brachydactylies (BDs) are a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. They have been classified on an anatomic and genetic basis into five groups, A to E, including three subgroups (A1 to A3) that usually manifest as autosomal dominant traits. BDA2 was described first in a large Norwegian kindred. BDA2 is caused by mutations in BMPR1B gene and studies demonstrate that these mutations function as dominant negatives in vitro and in vivo.
Function:
On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. Type II receptors phosphorylate and activate type I receptors which autophosphorylate, then bind and activate SMAD transcriptional regulators. Receptor for BMP7/OP-1 and GDF5.
Subcellular Location:
Membrane; Single-pass type I membrane protein.
DISEASE:
Defects in BMPR1B are the cause of acromesomelic chondrodysplasia with genital anomalies (AMDGA) [MIM:609441]. Acromesomelic chondrodysplasias are rare hereditary skeletal disorders characterized by short stature, very short limbs, and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers).
Defects in BMPR1B are a cause of brachydactyly type A2 (BDA2) [MIM:112600]. Brachydactylies (BDs) are a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. They have been classified on an anatomic and genetic basis into five groups, A to E, including three subgroups (A1 to A3) that usually manifest as autosomal dominant traits. BDA2 was described first in a large Norwegian kindred. BDA2 is caused by mutations in BMPR1B gene and studies demonstrate that these mutations function as dominant negatives in vitro and in vivo.
Similarity:
Belongs to the protein kinase superfamily. TKL Ser/Thr protein kinase family. TGFB receptor subfamily.
Contains 1 GS domain.
Contains 1 protein kinase domain.
SWISS:
O00238
Gene ID:
658
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
产品图片